
DEFINITION DE LA DM1
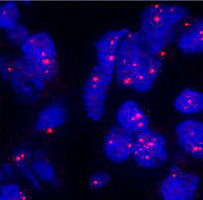 Agrégats d'ARN "mutés" (en rose) dans des noyaus de cellules cardiaques (avec l'aimable autorisation de G. Gourdon, Inserm)
Agrégats d'ARN "mutés" (en rose) dans des noyaus de cellules cardiaques (avec l'aimable autorisation de G. Gourdon, Inserm)
La dystrophie myotonique de type1 (DM1), appelée aussi maladie de Steinert ou myopathie de Steinert, est la plus fréquente des maladies neuromusculaires chez l’adulte. Il y aurait en France de 6 à 8000 personnes atteintes. En Europe, pour des raisons inconnues, la prévalence est plus élevée en Suède et au Pays Basque.
C’est une maladie génétique héréditaire, transmise de génération en génération par un parent porteur (lui-même descendant d’un parent déjà porteur ou chez qui l’anomalie génétique apparaît brutalement pour la première fois).
Elle affecte autant les individus de sexe féminin que de sexe masculin. Elle est transmise sur le mode « autosomique dominant », c'est-à-dire que chaque enfant d’un couple dont un des deux parents (homme ou femme) est atteint a une probabilité d’un sur deux d’être atteint lui-même.
En général elle s’aggrave à chaque génération (phénomène dit « d’anticipation ») : apparition de plus en plus précoce, symptômes plus nombreux et plus importants.
Comme toutes les myopathies, la maladie de Steinert se caractérise par des atteintes des muscles (affaiblissement des muscles appelé « dystrophie », troubles du tonus musculaire appelés « myotonie »), mais aussi par des dérèglements d’autres organes (appareil cardiorespiratoire, système digestif, yeux, système hormonal, système nerveux, …). Pour ces raisons, elle est dite « multi systémique ». En général, les atteintes s’aggravent plus ou moins rapidement avec l’âge (maladie évolutive). Cependant, les symptômes et leur évolution étant très variables suivant les individus, il n’est pas possible de prévoir l’évolution de la maladie chez un malade particulier.
La maladie est due à une anomalie qui se produit pour la première fois (pour des raisons encore inconnues) lors de la formation d’un embryon dans une génération d’individus. L’anomalie apparaît sur le chromosome 19 (les chromosomes portent sous forme d’ADN le patrimoine génétique de chaque individu, sorte de « livre de recette » qui sert de référence pour la fabrication de tous les éléments de l’organisme). Dans une certaine séquence de l’ADN de ce chromosome, à l’emplacement dit « 19q13-2.3 », se trouve le gène « DMPK » (pour « Dystrophie Myotonine Protéine Kinase ») codant une protéine qui intervient entre autres dans le fonctionnement du muscle. Dans le cas de la maladie, la séquence de codage est faussée par une répétition en surnombre d’un des « mots » du code (dit « triplet CTG »), qui, au lieu d’être répété au maximum 37 fois comme chez un individu sain, est « amplifiée » plus de 50 fois, pour atteindre quelquefois plusieurs milliers de répétitions
(3000 à 4000 répétitions CTG).
Entre 38 et 49 répétitions, on parle de « Pré-mutation » : il n’y a pas de symptômes, mais un risque de transmission avec augmentation des répétitions ; cependant, ce nombre peut rester stable durant plusieurs générations (en particulier si transmission par la mère).
Le diagnostic dans une famille est souvent fait parce qu’un enfant est atteint et présente des symptômes visibles.
Les principal mécanisme perturbateur identifié à l’heure actuelle est le suivant : lors de la recopie de l’ADN, cette « erreur de code » se propage aux transmetteurs de recopie de l’ADN (les « ARN messagers ») qui deviennent inopérants, ne peuvent pas sortir du noyau de la cellule pour aller piloter la synthèse des protéines concernées, s’accumulent dans le noyau en formant des agrégats, ce qui provoque des perturbations empêchant d’autres protéines d’être synthétisées à leur tour (d’où les atteintes diverses dans l’organisme).
En fait, nombreuses modifications sont avérées dans le transcriptome (ensemble des molécules, ARN, etc. issues de la transcription de l’ADN) des patients, ce qui entraîne des perturbations dans les processus d’épissage. Non seulement ces changements dépendent des facteurs de régulation comme MBNL, CUGBP, ETR3, mais également d’une multitude d’autres processus.
Notamment, les ARN toxiques « collent » les protéines d’épissage d’autre gènes, avec un effet « boule de neige » : tout va de plus en plus mal (épissage = processus de transformation de l’ARN messager en gène codant, par exclusion de « morceaux inutiles », les « introns »).
On sait par exemple que la perte de fonction des facteurs de régulation de l’épissage MBNL1 et MBNL2 est à l’origine de multiples perturbations : protéine Tau dans le cerveau, persistance anormale de structures de polyadénylations fœtales dans les tissus, épissage du canal sodium cardiaque SCN5A, expression anormale de l’exon 7 de la protéine « Bridging Integrator-1 » (BIN1) qui intervient dans la biogenèse des muscles (contribuerait à l’atrophie et à la faiblesse des muscles). L’épissage anormal de l’exon DMD 78 perturbe aussi la maintenance des fibres musculaires et contribue au processus d’évolution de la dystrophie. Il est d'ailleurs confirmé que les défauts d’épissage sont corrélés avec la faiblesse musculaire.
On sait également que les protéines de réparations d’erreurs MSH2 et MSH3 (qui maintiennent l’intégrité du génome = système de réparation automatique de l’ADN) sont impliquées dans la formation des répétitions CTG (elles rajouteraient des répétitions au lieu d’en supprimer).
D’autres mécanismes seraient également à l’œuvre : les répétitions en surnombre perturberaient aussi les gènes situés au voisinage de 19q13, codants pour d’autres protéines
C’est l’ensemble de ces désordres qui provoque l’aspect multisystèmique des symptômes de la maladie.
Le nombre des répétitions varie d’un individu à l’autre, varie dans les différents organes d’un même individu, évolue au cours de la vie du malade. Sauf en de rares exceptions, le nombre de répétitions augmente à chaque génération, c’est le phénomène dit « d’anticipation ». Il est établi que, statistiquement, la gravité et la précocité d’apparition des symptômes sont corrélées au nombre de répétitions. Cependant ce lien n’est pas valable au plan individuel, par exemple des personnes avec « seulement » quelques centaines de répétitions peuvent avoir des symptômes très graves et inversement.
La progression de la dynamique des mutations n’est ni linéaire, ni proportionnelle à la taille de l’allèle et variable dans le temps ; son comportement est plus ou moins rapide durant la vie du patient.
LES « FORMES » DE LA MALADIE
Bien que l’origine génétique soit la même, la gravité, le début d’apparition et l’évolution des symptômes sont différents suivant l’âge où apparaissent les symptômes et leur gravité. Un classement en quatre « formes » a été établi, qui vont de formes bénignes tardives jusqu’à des formes graves à la naissance.
- La forme asymptomatique. Les personnes ne s’aperçoivent pas des symptômes qui sont extrêmement légers et tardifs (par exemple calvitie ou cataracte), signes qui ne sont pas ressentis comme liés à une pathologie ; ces formes ne sont donc généralement diagnostiquées que lors de la recherche des origines génétiques d’un parent plus gravement atteint (frère, sœur, enfant, petit-enfant, cousin, neveu, ...).
- La forme adulte commune. Les symptômes apparaissent de façon légère vers 20-25 ans, mais le diagnostic est généralement posé vers 40 ans quand les symptômes s’aggravent (notamment la gêne dans la marche) ; ils évoluent plus ou moins rapidement suivant les individus ; chez les femmes, la grossesse peut être un facteur aggravant, voire déclenchant l’apparition des premiers symptômes.
- La forme infantile. Dans cette forme, on note certaines atteintes du système nerveux central, entraînant retard du langage (essentiellement en écriture et lecture) et de nombreuses difficultés scolaires par syndrome dysexécutif. Les enfants présentent des signes de fatigue, de lenteur, d’hypersomnie. Des signes autistiques apparaissent dans 1/3 des cas. En général, la myotonie et les problèmes musculaires arrivent plus tard. Des difficultés d’insertion professionnelle peuvent survenir.
- La forme néonatale (dite aussi congénitale). Transmise quasi-exclusivement par la mère (alors que les autres formes sont transmises indifféremment par le père ou par la mère), cette forme est très grave, avec hypotonie (bébé « mou »), détresse respiratoire à la naissance entraînant une mortalité néonatale élevée (plus de 15%) et difficulté à téter. Cette forme s’accompagne d’atteintes du système nerveux central (SNC), avec par la suite des signes plus ou moins importants de déficience mentale, un retard sévère d’acquisition du langage, des problèmes psychologiques. Des signes peuvent être détectés pendant la grossesse, comme un excès de liquide amniotique (hydramnios) ou des diminutions des mouvements du fœtus ; les risques d’accouchement prématuré ou difficile (présentation par le siège) sont particulièrement élevés. Les cas de survenue sont d’environ 10% en cas de première naissance, mais, pour des raisons non élucidées à ce jour, passent à 40% si une première naissance d’enfant atteint a déjà eu lieu.
Bien noter qu'il s'agit bien de la même maladie (anomalie du gène DMPK) et non de 4 maladies différentes ; d'ailleurs la frontière entre les 4 "formes" n'est pas rigide :
- d'une part, il y un continuum dans l'expression des symptômes, on parle par exemple de la forme « juvénile » (continuum infantile/adulte) et de la forme « tardive » (continuum adulte/asymptomatique)
- d'autre part, l'expression est extrêmement variable d'un individu à l'autre dans une même forme.
La répartition statistique générale entre les différentes formes dans la population atteinte est d’environ : 5% congénitale, 19 % infantile, 26 % juvénile, 39 % adulte, 11 % tardive.
En tout état de cause, l’analyse de l’ADN (consultation médicale de génétique) permettra de poser un diagnostic précis (avec des règles légales et déontologiques très strictes concernant cet examen chez les enfants).
Enfin, il faut noter que la maladie de Steinert n’est pas contagieuse et que personne n'est à blâmer pour son apparition initiale dans une lignée généalogique. On ne connaît pas à l’heure actuelle le mécanisme déclencheur de la mutation génétique.
SYMPTOMES & PRISE EN CHARGE
PRECAUTIONS ESSENTIELLES
Surveillance cardiaque régulière (au moins une fois par an)
Précautions spéciales en cas d’anesthésie (avertir l’anesthésiste)
Conseil génétique pour la procréation
Porter sur soi la carte de soins et d’urgences
Le médecin est seul habilité à prescrire la prise en charge la mieux adaptée à chaque patient. Les prises en charge décrites sont données à titre informatif comme étant les plus fréquentes et ne sauraient tenir lieu de prescription.
Noter que dans le cas de la maladie de Steinert, les traitements prescrits sont en général pris en charge à 100% par l’assurance maladie.
Mise à jour Février 2025
Site créé le 20 octobre 2013